Directed Evolution

We mimic natural selection to develop proteins with novel and advanced properties. Through iterative rounds of mutation and screening/selection, we traverse fitness landscapes to find optimal proteins for user defined goals.
Directed evolution circumvents our profound ignorance of how a protein’s sequence encodes its function by using iterative rounds of random mutation and artificial selection to discover new and useful proteins. Proteins can be tuned to adapt to new functions or environments by simple adaptive walks involving small numbers of mutations. Directed evolution studies have shown how rapidly some proteins can evolve under strong selection pressures and, because the entire ‘fossil record’ of evolutionary intermediates is available for detailed study, they have provided new insight into the relationship between sequence and function. Directed evolution has also shown how mutations that are functionally neutral can set the stage for further adaptation.
How to Create New Enzyme Functions in the Laboratory
For the most part, directed evolution is conceptually and technically straightforward: once an enzyme that displays a low level of activity for a desired function is identified, mutagenesis and screening for improved function will often provide enhancements. However, that first step—identifying a protein with some initial activity—can be far from straightforward. How does a protein that performs one function evolve into another with a different function? And how can enzyme engineers accomplish this quickly to address current, time-sensitive problems? Two key concepts for accomplishing this are catalytic promiscuity—the ability of an enzyme to carry out functions other than its primary one—and chemical intuition—the how and why a reaction might happen.
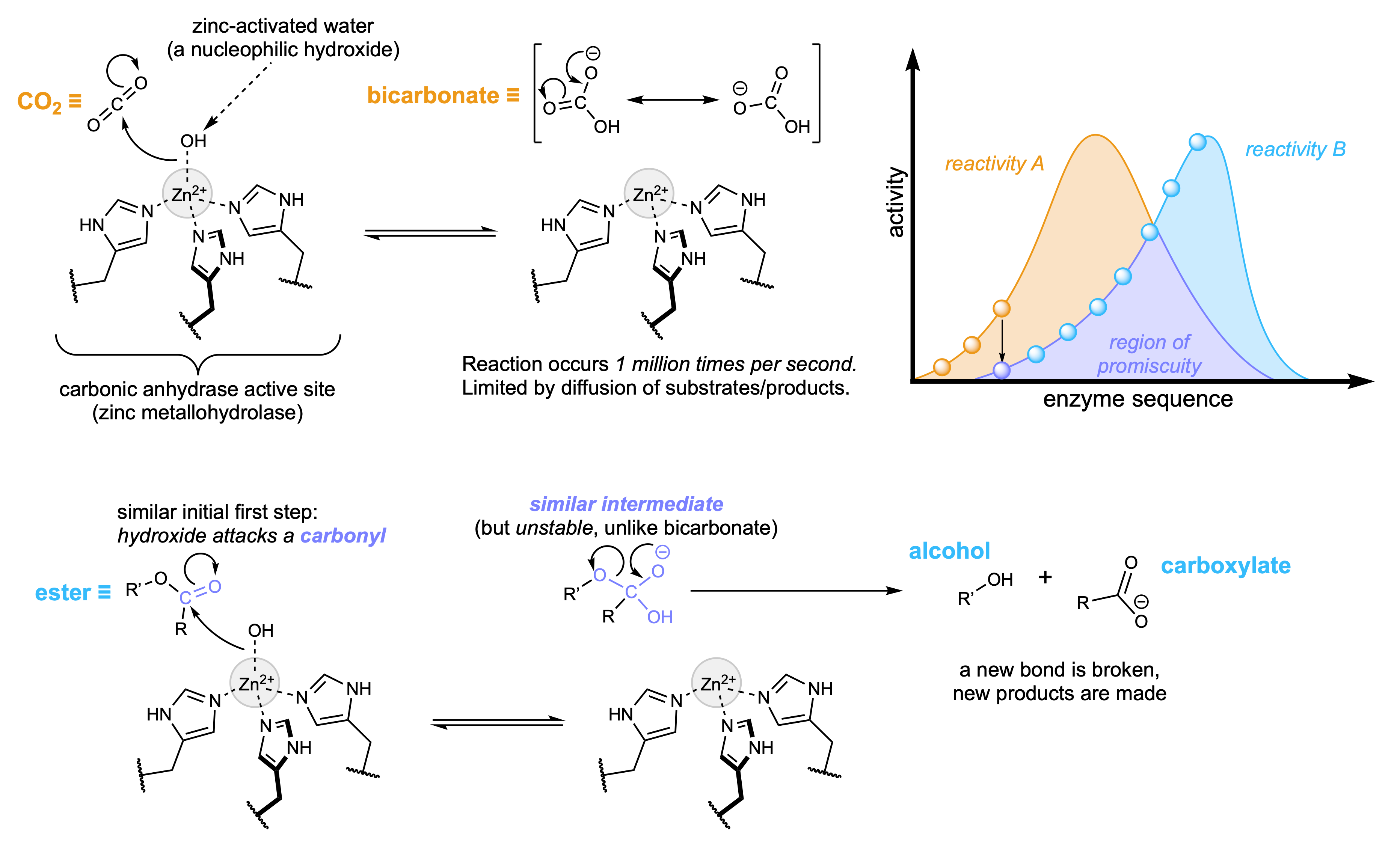
When looking to create a new enzyme for a reaction that is not known to be catalyzed by an existing enzyme, we must hypothesize how a new reaction might be catalyzed, find protein(s) that might be able to do that, and test them using the appropriate substrates (and sometimes even cofactors). This process can guide us to identify a starting point for directed evolution of a new enzyme function. It may even happen over multiple directed evolution campaigns, iteratively accessing new activities as others are optimized.
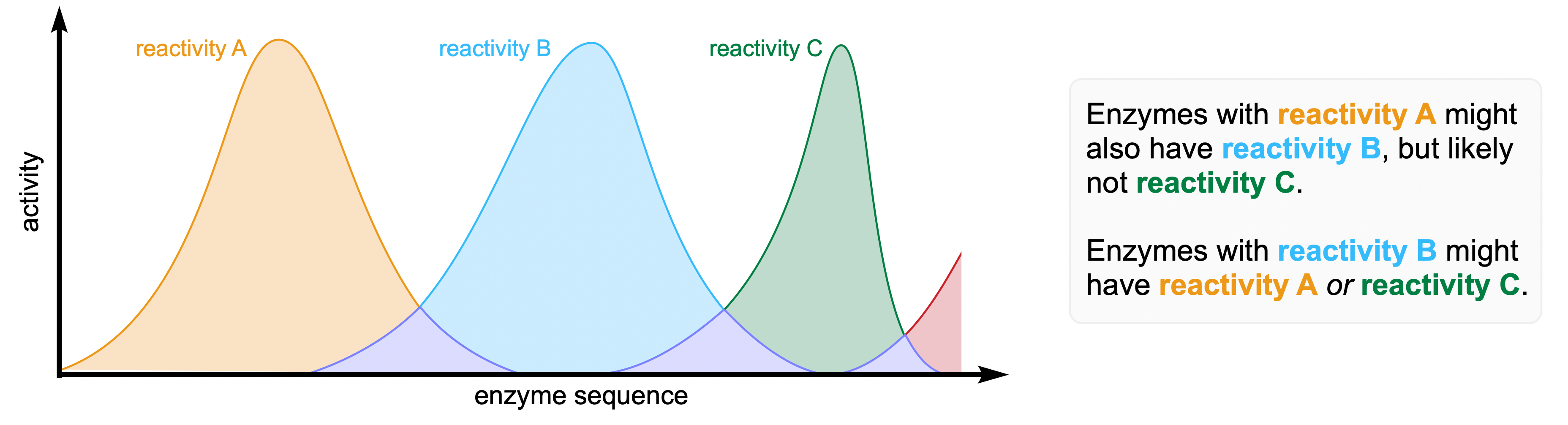
Read more about how to leverage chemical intuition and enzyme promiscuity to create new enzyme functions here.
Crystallography
Crystallography enables structural characterization of proteins, providing molecular insights and guiding protein design.
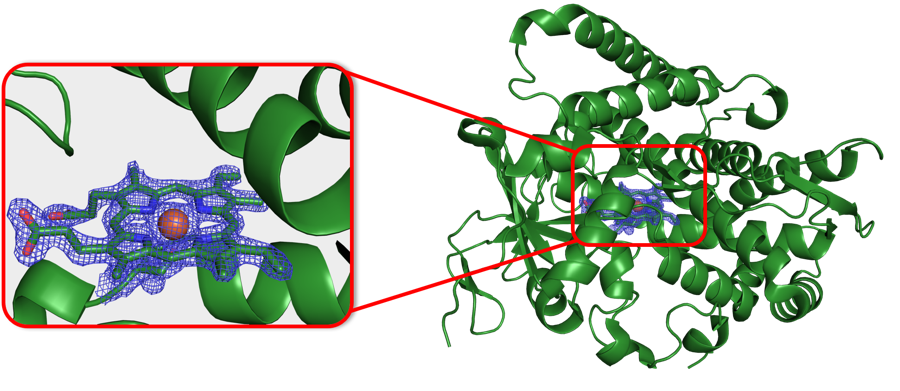
We use x-ray crystallography to structurally characterize the proteins we have engineered. We can visualize protein-subunit interfaces involved in activity regulation, active site organization of our enzymes, and substrate and cofactor binding-sites. Visualizing our advanced protein variants at the molecular level tells the story behind beneficial mutations. These crystal structures provide the foundation of our protein design efforts.
Check out our structures!
Every Variant Sequencing
We have developed cost-effective every variant sequencing technology.

Sequence-function data provides valuable information about the protein functional landscape but is rarely obtained during directed evolution campaigns. We first developed every variant sequencing (evSeq), and recently developed Long-read every variant Sequencing (LevSeq), a pipeline that combines a dual barcoding strategy with nanopore sequencing to rapidly generate sequence-function data for entire protein-coding genes.
Check out our codebase for LevSeq pipeline, visualization and website as well as the codebase for evSeq!
Machine-Learning Assisted Protein Engineering
We have developed various machine learning-based methods to enable more efficient initial activity discovery and protein engineering.

Machine learning-assisted directed evolution (MLDE) enables more efficient protein engineering by learning the relationship between protein sequences and fitness (numerical metrics for protein function) from small training (experimental) datasets to predict high-fitness variants across the sequence landscape. Since our original development of MLDE, we introduced active learning-assisted directed evolution (ALDE), and focused training. We also provided tools for
evaluating variations of these MLDE strategies.
Check out our codebase ALDE pipeline, focused training and method evaluation.